Myotonic Dystrophy
Stem:
- This patient has presented with muscle weakness, please examine his neurological system
- Shake hand: grip myotonia, whereby there is continued contraction of muscle after voluntary contraction ceases, following by impaired relaxation. But grip myotonia can be absent with advanced disease as wasting of the small muscles of the hands progresses
- General examination
- Myopathic facies - lifeless, lean and expressionless face with bilateral ptosis and flaccid muscles around the mouth and eye. General drooping appearance of the moth, sometimes hanging open
- Pt may look tired due to daytime somnolence as a consequence of nocturnal hypoventilation
- Frontal balding
- Cataracts: usually posterior sub-capsular in origin. Look for evidence of previous cataract surgery
- Hands
- Myotonic dystrophy is a distal myopathy - wasting of the small muscles of the hands with dorsal guttering
- Percussion myotonia can be elicited by percussion at the hypothenar eminence which leaves a dent and fills slowly as a result of myotonia. Note to illicit percussion myotonia one has to tap with a reflex tendon hammer on the thenar eminence.
- Feet
- Bilateral foot drop with high steppage gait
- Reflexes
- deep tendon reflexes may be depressed or lost
- Facial features
- General examination as described above
- Demonstrate facial weakness - easily overcoming eye closure, perioral weakness
- Eyes - cataracts
- Speech
- dysarthria, slow and low volume speech
- Other relevant systems:
- Gastrointestinal - colicky abdominal pain, constipation, diarrhea, and pseudo-obstruction due to smooth muscle involvement
- Cardiovascular - cardiomyopathy, heart failure, conduction disorders, and arrhythmias
- Atrioventricular and intraventricular conduction disturbances are the most frequent abnormalities
- Structural heart abnormalities have also been associated - LV hypertrophy, dysfunction, dilatation
- Primary hypogonadism (low-serum testosterone, elevated serum FSH concentration, oligospermia, and infertility), testicular atrophy, and associated oligospermia or azoospermia with infertility are common problems in DM1
- Endocrine - insulin resistance and thyroid nodules may be present
What is the differential diagnosis of bilateral ptosis?
- Myasthenia gravis
- Kearns Sayre syndrome
- Fascioscapulohumeral muscular dystrophy
- oculopharyngeal muscular dystrophy - can be either autosomal dominant in inheritance or autosomal recessive, the later is rarer. It is caused by mutations that lead to increased alanine levels in the protein PABPN1, which is involved in stabilising mRNA within cells. The abnormal protein accumulates inside the nucleus, causing muscle cell loss. Compared to myotonic dystrophy, oculopharyngeal muscular dystrophy causes a proximal myopathy in comparison to a distal myopathy (small hands affected by myotonic dystrophy)
What is the pathophysiology of myotonic dystrophy?
- Multisystem disease
- There are two types, myotonic dystrophy type 1 and myotonic dystrophy type 2
- Genetics:
- Myotonic dystrophy type 1 - autosomal dominant - CTG trinucleotide expansion in myotonin protein kinase gene chromosome 19
- Myotonic dystrophy type 2 - expanded tetranucleotide repeat is located within the first intron of the ZNF9 (CNBP) gene, chromosome 3.
How do you differentiate between myotonic dystrophy type 1 and type 2?
- Genetics
- MDT1 - trinucleotide repeat chromosome 19
- MDT2 - tetranucleotide repeat chromosome 3
- Skeletal muscle weakness
- MDT2 - weakness occurs more proximally - weakness in the hip girdle region is often the presenting feature
- In general, MDT2 is less severe in its manifestations
Spastic (+/- hereditary) Paraparesis
General inspection
How would you investigate this patient?
What is the difference between spasticity and rigidity?
What is your differential diangosis of a patient who presents with bilateral spastic paraparesis
- May have prominent lower limb wasting secondary to disuse atrophy
- Scar on the back to suggest spinal surgery secondary to trauma or tumour
- Contractures - there may be scars over the tendons that represent corrective surgery for contractures and deformities
- Scissor gait
- Increased tone
- Presence of ankle clonus
- look for pyramidal distribution of weakness - which in the lower limbs is weakness of the flexor muscle groups
- Lower motor neuron type weakness if motor neuron disease suspected
- Hyper-reflexia
- Extensor plantar response which is evidenced by big tow extension, fanning of all toes or involuntary contraction of leg flexors
- Presence of a sensory level
- In hereditary spastic paraparesis there is no sensory signs
How would you investigate this patient?
- Perform a full neurological examination looking for other signs eg of motor neuron disease or multiple sclerosis
- Blood tests: B12/ folate for subacute combined degeneration of the spine, syphilis serology for tabes dorsalis
- MRI of the spine and brain
What is the difference between spasticity and rigidity?
- Spasticity is a velocity dependent increase in tonic stretch reflexes due to hyper-excitability whereas rigidity is present throughout the whole range of movement
- The pathophysiology of spasticity can be explained by unhindered re-inforcement of the muscle stretch reflex, which normally has tonic descending inhibition to the spinal alpha motor neurons
What is your differential diangosis of a patient who presents with bilateral spastic paraparesis
- For all neurological conditions think about where the lesion is likely to be and the aetiology. A good way to sort out the aetiology is through using the acronym VINDICATE (Vascular, Inflammatory, neoplastic, degenerative/ deficiency, idiopathic/ infectious, congenital, auto-immune/ allergy, traumatic, endocrine)
- Vascular - stroke, anterior spinal artery syndrome, Inflammatory - demyelinating lesions/ vasculitic lesions with secondary cerebral damage, neoplasms, degenerative/ deficiency (B12, leukodystrophies), idiopathic, infectious (tabes dorsalis, Tropical spastic paraparesis caused by human T-lymphotropic virus 1), congenital (hereditary spastic paralysis), autoimmune (MS, atypical neuromyelitis optica), Traumatic, Endocrine (not really)
Myasthenia Gravis
Usually the stem would be - please examine the face and proceed.
General inspection:
General inspection:
- Hooked up to intragram infusion
- Evidence of bilateral ptosis secondary to fatiguability - although often unilateral, and if bilateral it is asymmetrical
- May have a scar secondary to excision of a thymoma
- Essential to demonstrate to the examiner you are looking for fatiguability
- Weakness upon sustained up-gaze with furrowing of eye as a compensation (with use of frontalis mm), this is made better by resting the eyelid by closure for a short period of time
- Extra-occular mm weakness - test full range of movement and assess for diplopia. Use the cover test to decide which mm is affected.
- Peek sign: Close eyes and within 30s the eyes start to peek open
- Horizontal smile, known as the myasthenic snarl
- Weakness of mouth opening/ closing. In MG, the mouth closure mm tend to be more affected. Fatiguability may be tested
- Jaw supporting sign - pathognomonic of MG, may appear later in clinical examination
- Weakness of palatal mm - nasal quality of speech. Ask patient to count progressively and assess for change in the quality of the speech
- Test neck weakness - neck flexors are more affected than neck extensors
- Neck scar secondary to thymectomy
- Prox limb girdle mm - repeated Shoulder Abdxn/ Addxn. Tested by first checking abduction together. Then rest one shoulder and repeat abduction/ adduction 15 times, then re-test both shoulders to demonstrate fatiguability
- Sensation should be normal, reflexes should be normal, as these do not involve the neuromuscular junction consisting of nicotinic AChR
- For signs of immunosupression
- For signs of vascular access - eg for plasma exchange therapy
- Look for other associated autoimmune conditions - hyperthyroidism, hypothyroidism, diabetes, addisons disease, pernicious anaemia, pemphigus, chronic active autoimmune hepatitis
- Myopathy - eg fascioscapulohumeral muscular dystrophy, myotonic dystrophy, oculopharyngeal muscular dystrophy, mitochondrial myopathy
- Neuromuscular junction pathology - lambert-eaton myasthenic syndrome, botulism
- Individual neuropathies (mononeuritis multiplex, extrinsic compression, infiltrative process), miller-fischer syndrome
- Anterior horn cell pathology (motor neuron disease)
- CNS pathology: Vascular insult, mass lesion, demyelinating pathology, leptomeningeal disease)
- Tensilon test - need IV access, inject placebo (normal saline) and test fatiguability, subsequently inject edrophonium, a reversible anticholinesterase inhibitor whilst they are on cardiac monitoring and determine if their weakness improves.
- Bloods: nACHr abs (found in 80 - 90% of pts), anti-muscle specific kinase (MuSK) antibodies, voltage gated Calcium channels (For LEMS)
- CT-thorax: Looking for thymoma
- EMG Repetitive stimulation → progressive ↓amplitude mm APs
- Needle examination of affected mm shows motor unit potential variation & sometimes fibrillation potentials & myopathic change
- Single-fibre EMG → ↑d jitter & blocking
- TFTs, RF & ANAs – to screen for associated Autoimmune conditions
- PFTs if suggestion of Resp mm involvement
- Two types of myasthenia: Ocular myasthenia --> 15%, limited to eyelids and EOMs and Generalised myasthenia --> 85%
- MuSK is important in clustering of nAChR on post-synaptic membrane on NMJ, is found on sero-negative myasthenia gravis, more generalised with bulbar involvement and respiratory weakness
- Drugs can exacerbate MG --> aminoglycosides, penicillamine, fluroquinolones, macrolides
- 15% of patients have thyomoma, invasive disease is rare. 65% have thymic hyperplasia. Pt with thymoma have more severe and generalised MG, with higher disease titres. The antibody is pathogenic.
- Confirm diagnosis with serology, tensilon test, repetitive nerve stimulation studies, single fibre EMG recordings
- Optimise symptoms: acetylcholinesterase inhibitors, stop drugs that may trigger or exacerbate MG such as penicillamine, aminoglycosides, fluroquinolones
- Prevent deterioration: steroids if severe disease, move on to immunosuppressive therapy such as azathioprine, mycophenylate, cyclosporin, thymectomy if sero+ and generalised, 70% show improvement, 25% of those → remission
- Manage Exacerbations: Eg myasthenic crisis --> HDU monitoring, monitor vital capacity, SC/ IV Neostigmine, plasmapheresis (note fatigue is a side effect)
Cerebellar syndromes
Clinical signs of cerebellar syndrome:
What are the causes of a cerebellar syndrome?
- Gait
- wide based gait
- mild cerebellar gait may be manifest as difficulty in tandem walking. Test this as this may not otherwise be elicited
- Romberg's sign occurs when patients lose balance after closing their eyes due to either vestibular dysfunction or loss of proprioception
- truncal ataxia in midline (vermis, floculonocular lobe) lesion
- Upper limbs
- Rebound phenomenon - also known as the holmes rebound phenomenon
- Hypotonia - associated with lateral cerebellar lesions
- Intention tremor - tremor that begins and increases as patient reaches the target
- dyssynergia - incoordination, where patient lacks smoothness of execution of various motor activities
- dysmetria - inability to control range of movement, leading to overshooting of target
- Dysdiadochokinesis - form of limb ataxia
- Cranial nerves
- abnormal posture of head - may be due to midline or lateral cerebellar lesion
- Nystagmus - horizontal nystagmus (75%), fast phase towards the side of the lesion
- Ocular dysmetria - hypermetric saccades
- opsoclonus - constant, random, conjugate sacades of unequal amplitudes in all directions
- dysarthria - slow, laboured, slurred speech, staccato like, scanning speech - "british constitution, irish constabulatory"
- Lower limbs
- impaired heel-shin testing
- pendular knee jerk as a result of hypotonia
What are the causes of a cerebellar syndrome?
- Vascular causes - posterior circulation ischaemia, lateral medullary syndrome, cerebral vasculitis
- Infective - cerebral abscess, TB, toxoplasmosis
- Traumatic/ Toxins - lithium, alcohol, phenytoin, carbemazepine
- Auto-immune - demyelination (MS), SLE
- Metabolic - hypothyroidism
- Inherited - Spinocerebellar ataxias, Friedrich's ataxia
- Neoplastic - posterior fossa tumours such as medulloblastoma's, astrocytomas, paraneoplastic phenomenon from small cel lung cancer, breast and ovarian cancer with anti-purkinje cell anti-bodies
- Degenerative - MSA (cerebellar subtype)
- Congenital - bud-chiari malformation
Peripheral Neuropathy
General principles:
What are the causes of a axonopathy and what are its characteristics?
What are the 6 different patterns of diabetic neuropathy?
(1) distal symmetric sensorimotor axonal neuropathy
(2) distal predominantly small fibre neuropathy
(3) autonomic neuropathy
(4) proximal neuropathy [femoral amyotrophy, diabetic lumbosacral radiculoplexus neurpathy]
(5) mononeuritis multiplex
(6) entrapment neuropathies
What are the causes of a ganglionopathy?
What toxins are associated with an axonopathy?
What is the typical pattern of a demyelinating peripheral neuropathy?
What is the differential diagnosis for a demyelinating predominant neuropathy?
What infections have been associated with acute inflammatory demyelinating polyneuropathy?
What are the clinical features of AIDP?
How would you diagnose and manage AIDP?
How do you distinguish CIDP from AIDP?
What are the hereditary neuropathies to be aware of?
- Very common short case as there are a lot of patients present
- Nerves subdivided into cell bodies (neuronopathy), axons (axonopathy) and myelin sheaths (myelinopathy)
- Motor fibres are carried by large myelinated fibres
- Vibration and proprioception is carried by large myelinated fibres (A-beta)
- Pin-prick and temperature carried by small myelinated (a-delta) and unmyelinated (c fibres) fibres
- Aids to walking, scuffed shoes, orthosis
- High steppage gait due to proprioceptive loss
- Thickened nerves in amyloid, leprosy, hereditary motor and sensory neuropathy
- Maculoanesthetic patches in leprosy
- Mee’s line on the nail with arsenic poisoning
- Orange tonsils in tangier’s disease
- Loss of leg hair, calluses, ulceration, deformities such as pes cavus and high arched feet
- Areas of warmth, oedema, erythema in autonomic neuropathy. Also patient may have indwelling catheter in situ
- Gait:
- Rhomberg’s positive if proprioceptive loss
- High steppage gait if there is presence of foot drop
- Tone normal
- Power
- largely unaffected if predominantly sensory: causes include diabetes, uraemia, drugs {isoniazid, colchicine, amiodarone, allopurinol, cisplatin, vincristine, taxanes, gold), alcohol, thiamine deficiency, hereditary (CMT, amyloidosis), sarcoidosis, hypothyroidism, vasculitis, B12 deficiency, copper deficiency
- If distal > proximal consider causes of axonopathy: axonal variant of CIDP, Multifocal axonal demyelinating sensory and motor neuropathy (MADSAM)
- If proximal > distal consider demyelinating pathology (GBS, CIDP, multifocal neuropathy, paraproteinemic neuropathy, POEMS, HIV, drugs such as amiodarone, bortezomib, TNF-alpha)
- If asymmetric consider polyradiculopathy, plexopathy secondary to DM, meningeal carcinomatosis or lymphomatosis
- Reflexes
- Tend to be areflexic
- Because either the afferent or efferent reflex arc has been involved
- Ankle jerk reflexes are generally the first to be lost
- Sensation
- Axonopathies typically present with distal loss of sensation
- Large fibre loss occurs in context of diabetes, alcohol, B12, vitamin E, copper
- Ganglionopathies can cause severe sensory ataxia and impairments in proprioception
- Vitamin B6 toxicity can cause ganglionopathy
- When level of stocking distribution above knee, then there will also be sensory loss at fingers from axonopathy
What are the causes of a axonopathy and what are its characteristics?
- Axonopathy typically causes sensory loss first, then followed by motor loss. Reflexes are involved early
- The typical pattern of an axonopathy is length dependent
- Causes of an axonopathy can be remembered by the following mnenonic:
- DAT-PUNC-V
- Diabetes, alcohol, toxins (Pb, arsenic, thallium etc), paraneoplastic (voltage gated potassium channel, anti-hu), uraemic, nutritional (B12 deficiency, B6 toxicity), critical illness, vasculitis
What are the 6 different patterns of diabetic neuropathy?
(1) distal symmetric sensorimotor axonal neuropathy
(2) distal predominantly small fibre neuropathy
(3) autonomic neuropathy
(4) proximal neuropathy [femoral amyotrophy, diabetic lumbosacral radiculoplexus neurpathy]
(5) mononeuritis multiplex
(6) entrapment neuropathies
What are the causes of a ganglionopathy?
- The symptoms of a ganglionopathy include predominant sensory ataxia, proprioceptive loss
- Pseudoathetosis may also occur
- All sensory modalities can be affected as their cell bodies lie within the dorsal root ganglion
- Causes include anti-Hu antibody (small cell paraneoplastic phenomenon), anti-FGFR3 antibody sensory neuronopathy, associated with sjogren's and lupus, sjogren's syndrome, idiopathic sensory neuronopathy
What toxins are associated with an axonopathy?
- chemotherapy related include platinum compounds, taxanes, bortezomib; chloroquine; B6 toxicity, Isoniazid, colchicine, amiodarone, thalidomide, lenalidomide, lead = basophilic stippling, predominantly upper limbs
What is the typical pattern of a demyelinating peripheral neuropathy?
- Whilst there are no absolute rules, a demyelinating peripheral neuropathy typically affects motor > sensory fibres and there is weakness out of proportion to wasting
What is the differential diagnosis for a demyelinating predominant neuropathy?
- Mnemonic: MIIHD
- Metabolic & Malignant [storage diseases/ mitochondrial/ paraproteinemic/ POEMS]/ Immune [AIDP/ CIDP]/ Infectious [botulinum/ HIV]/ Drugs [amiodarone/ bortezomib/ TNF-alphas, hexa-carbons]/ Hereditary [Charcot-Marie-Tooth]
What infections have been associated with acute inflammatory demyelinating polyneuropathy?
- Most commonly C. jejuni, but also M. pneumoniae, CMV, EBV
- Adverse prognostic features include early need for ventilation, acute onset severe quadriparesis and small initial CMAPS implies axonal degeneration
What are the clinical features of AIDP?
- Ascending paralysis, early loss deep tendon reflexes, pain = early feature, CN involvement, progression complete by 4 weeks, recovery over weeks to months, variable degree sensory involvement
- Axonal GBS = acute motor axonal neuropathy, acute motor sensory axonal neuropathy: more severe, extensive neuronal loss
- Miller Fisher variant: ataxia, ophthalmoplegia, areflexia +/- bulbar/ facial weakness
- Autonomic features
How would you diagnose and manage AIDP?
- Clinical and laboratory features.
- Diagnostic tests include antibodies (eg anti GM1 for acute motor axonal neuropathy, anti-GQ1b for miller fisher variant)
- lumbar puncture (cyto-albuminaemic dissociation which is raised protein but normal cells in LP)
- nerve conduction studies (evidence of demyelination by conduction block, temporal dispersion, increased latency, F wave and H reflex is more sensitive as you get information more proximally)
- Decreased compound motor action potential is an adverse prognostic feature in AIDP as it implies axonal involvement
- Steroids have no role in the treatment of AIDP
- IVIG has similar efficacy to plasma exchange, and it also halts progression
How do you distinguish CIDP from AIDP?
- CIDP progresses over 8 weeks, whereas AIDP reaches a nadir by 4 weeks
- It is important to distinguish the two because CIDP remits with corticosteroids and immunosuppression
- Acute antibodies are not present as they are in AIDP
What are the hereditary neuropathies to be aware of?
- Hereditary Motor Sensory Neuropathy 1 = CMT1 = demyelinating, A/D, onion bulbs, hypertrophic
- Hereditary Motor Sensory Neuropathy 2 = CMT2 = axonal, A/D, no slowed velocities on nerve conduction
- Hereditary sensitivity to pressure palsies = A/D, mononeuropathy brought out by trivial compressions, can take weeks or months to resolve
Parkinson's disease and Parkinson's plus syndromes
General comments
What is the differential diagnosis of patients who present with parkinsonism?
What are the clinical features of multiple systems atrophy?
What are the clinical features of progressive supranuclear palsy?
What are the clinical features of corticobasal degeneration?
What are the clinical features of lewy body dementia?
Outline the management options in parkinsons disease
- The stem is usually please examine this patient's gait and proceed or "this patient has a tremor" please examine the upper limbs and proceed
- asymmetrical signs support parkinson's disease in contrast to symmetrical signs which support parkinsonism
- Walking aids
- Hypokinetic (mask-like, expressionless = hypomimia) facies - infrequent , drooling (due to dysphagia and autonomic dysfunction)
- Paucity of movement
- Resting tremor - coarse
- Improves w/ posture/intention
- To bring out you can ask the patient to perform mental arithmetic (ask patient to count backwards from 20) or ask the patient to move the contralateral limb "pretend you are painting the wall with the other arm". If the tremor is accentuated this is called synkinesis
- Dystonia - maybe medication related
- Speech: low volume, monotonous, tremulous, stutter, pallilalia (involuntary repetition of syllables, words, phrases)
- Difficulty initiating voluntary movements and starting to walk - freezing phenomenon
- Face: glabellar tap - this is not a specific sign, but tapping on the forehead should cause a patient with parkinson's disease to continue blinking
- Brow may be greasy with evidence of seborrhoea and seborrhoeic dermatitis due to abnormal swetting, a manifestation of autonomic dysfunction
- Posture stooped
- Bradykinesia
- Shuffling Gait, but in pure parkinson's disease the gait has a narrowed base. If there is a wide base think about MSA-cerebellar phenotype
- Reduced arm swing
- Festination
- Difficulty turning
- Loss of postural reflexes as manifest by propulsion (tendency to move forwards) and retropulsion (tendency to move backwards)
- Tone: cog-wheel or Lead pipe rigidity (reinforce by turning head side to side). Differentiate this from spasticity, which is increased tone at the initiation of movement which then characteristically gives way - a sign of an upper motor neuron lesion
- cog-wheeling should be demonstrated at the wrist through circular movements
- Tremor: resting, pill-rolling
- Finger-nose testing: tremor improves (as opposed to an intention tremor)
- Rigidity: Lead pipe and cog-wheel
- Bradykinesia
- Play piano on back of hand
- Form pincers with fingers
- Repeated duck bill movments
- Micrographia
- BP and Postural BP (autonomic dysfunction/MSA)
- Cerebellar tests: truncal ataxia (MSA)
What is the differential diagnosis of patients who present with parkinsonism?
- Vascular parkinsons disease
- Idiopathic parkinsons disease
- Lewy body dementia
- Parksinon's plus syndromes - multiple systems atrophy, progressive supranuclear palsy, corticobasal degeneration, lewy body dementia
- Drug induced: MPTP, dopaminergic blockers (typical antipsychotic agents such as haloperidol), metoclopramide, chlopromazine
- Basal ganglia pathology such as tumour
- normal pressure hydrocephalus
What are the clinical features of multiple systems atrophy?
- This is also known as shy-drager syndrome, and is an alpha-synucleopathy
- There are two predominant subtypes, those with parkinsonism (MSA-P) and those with cerebellar dysfunction (MSA-C)
- Classic features include camptocormia (severe anterior flexion of the spine) and disproportionate anterocollis
- Early falls
- Cerebellar features with wide based gait
- Early autonomic failure - Nearly all men with MSA develop early erectile dysfunction
- Respiratory stridor affects about 30% of patients at some stage and when combined with parkinsonism is highly suggestive of multiple system atrophy
- Hyper reflexia in conjunction with spasticity and a positive Babinski response is only seen in MSA and not IPD
- Imaging shows hot cross bun + putaminal rim sign, cerebellar degeneration if MSA-C
What are the clinical features of progressive supranuclear palsy?
- Progressive supranuclear palsy (PSP) presents as a symmetrical rather than asymmetrical Akinetic rigid syndrome in contrast to IPD
- It involves the trunk and neck rather than the limbs, causing early postural and gait instability
- Abnormal vertical then horizontal saccades
- Retrocolis may be feature
- Dysarthria, dysphagia, rigidity, frontal cognitive abnormalities, and sleep disturbances are additional common clinical features
- Asymmetrical parkinsonism
- MRI --> hummingbird sign resulting from significant atrophy of the midbrain with relative sparking of the pons
- This is a tauopathy
What are the clinical features of corticobasal degeneration?
- alien hand phenomenon, rigidity, dystonia, myoclonus, cortical sensory signs [neglect, dysgraphaesthesia, asterognosis, speech apraxis, tool substitution --> where brush teeth with fingers
- unlike other akinetic rigid syndromes, there is striking apraxia of the affected limb and the patient complains that the limb does not behave itself or follow orders and that it feels as if it belongs to somebody else.
- pathology --> tauopathy
- imaging --> cortical atrophy
What are the clinical features of lewy body dementia?
- Diffuse Lewy body disease presents with an akinetic rigid state in up to 40% of cases
- Approximately 90% will develop akinetic rigid syndrome in the course of illness
- other features are hypophonic speech, masked facies, stooped posture and festinating gait
- rest tremor is uncommon in DLBD
- There is presence of syncope, repeated falls and neuroleptic sensitivity in this disease
- DLBD is characterized by presence of cognitive deficits within a year of diagnosis as opposed to PD
- The dementia in DLBD is associated with prominent visual hallucinations and a fluctuating mental state
- REM sleep behavior disorders are also frequent in this disease though they are not specific for it
Outline the management options in parkinsons disease
- Non-pharmacological management: support groups:
- Education and support groups are important
- Speech therapy for hypomimia, monotonous, tremulous speech
- Physical therapy: Mounting evidence suggests that regular aerobic exercise has a positive impact on PD, tai chi twice weekly has RCT evidence in terms of stride length, functional reach etc. Also fewer self reported falls
- multidisciplinary rehabilitation including physical therapy and occupational therapy
- Pharmacological management
- L-DOPA with peripheral decarboxylase inhibitor = sinemet/ madopar --> no real difference if starting this or dopamine agonist or MOA-B, just start one of them. As per PD MED 2014 trial
- Wide therapeutic window at the start [motor fluctuations], then narrows in course as number of dopamine cells eventually fail, finally brain dopamine levels reflect serum levels --> take a pill switch on, pill wears off --> switch off
- If get dyskinesia --> option is to reduce dose or fractionate. Dyskinesia’s reflect reduced buffering of dopamine by SN cells
- If use in restless leg syndrome can get paradoxical augmentation therefore not first line in this setting
- Dopamine agonist --> pramipexole [D2/D3 specific] SE = impulse behaviour disorders
- MOAB --> rasagiline :prolongs action of L-dopa by decreasing metabolism [MAO involved in metabolism]
- Entacapone --> addition of COMT --> increases “ON” time, increased risk of dyskinesias
- Apomorphine injections/ infusions --> if controlling on-off symptoms, also used in patients who cannot take oral medications
- Duodopa infusions through PEG tube directly into duodenum
- Deep brain stimulation at the subthalamic nuclei or globus pallidus internus
- In young patients --> early deep brain stimulation, very small risk, intervene before lose too much quality of life.
- Management of psychosis in parkinson's disease
- all anti-parkinsonian drugs can produce confusion and hallucinations, therefore anticholinergic drugs should be withdrawn and L-DOPA dose should be reduced
- Best evidence exists for clozapine, but olnzapine and quetiapine may also be effective
Dysarthria and dysphasia
Dysarthria
Dysphasia
- Ataxic dysarthria
- This pattern occurs because there is a lack of cerebellar co-ordination of muscles of articulation, phonation and respiration
- The speech lacks modulation, is slurred and imprecise.
- There is distortions at the end of the words
- Rate of speech is variable, being explosive at certain times, and slow and scanning at other times
- may bite cheek and tongue whilst talking
- Spastic dysarthria
- Speech lacks modulation
- nasal quality
- imprecise articulation
- articulatory muscles are rigid in nature
- usually due to bilateral upper motor neuron lesions to cranial nerves involved in articulation (10 and 12)
- Hypokinetic dysarthria
- slow and monotonous speech
- no modulation
- low volume, low pitch
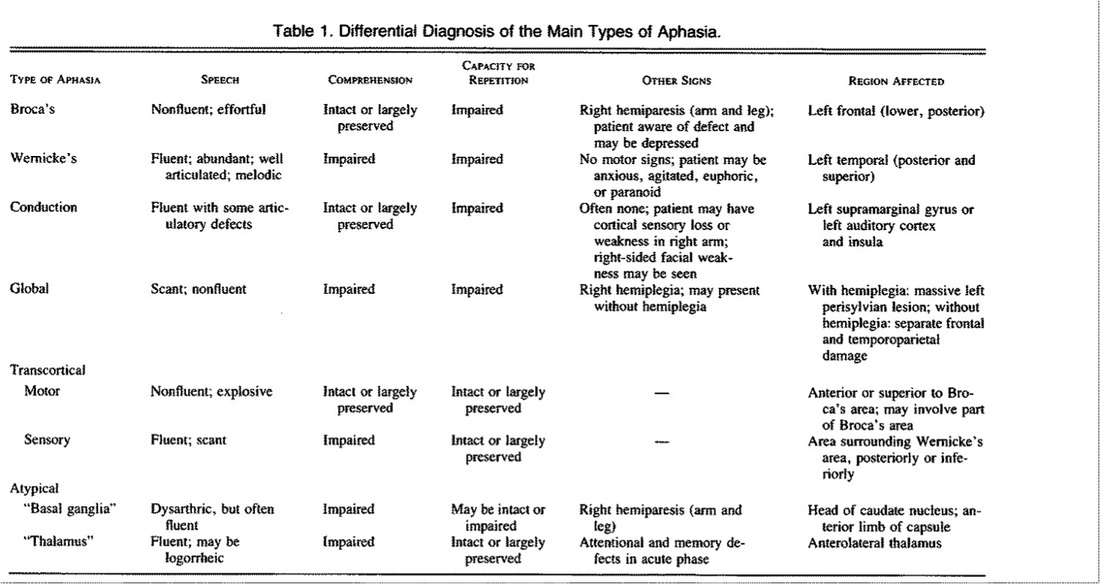
Dysphasia
- Characterise as fluent vs non fluent
- Determine if comprehension is impaired
- Determine if repetition is impaired
- Determine if naming is impaired
- Wernicke's aphasia
- This affects the posterior part of the superior temporal gyrus
- Speech is often fluent and with correct grammar
- There are often paraphasia and neologisms, and the speech may be incomprehensible
- Repetition is impaired, with paraphasias and neologisms
- Comprehension is impaired
- The patient often demonstrates anosognosia, or lack of awareness of the deficit
- Brocca's aphasia
- This affects he inferior left frontal gyrus, which contains the cytoarchitectonic fields [Brodmann's areas] 44 and 45
- drastic loss of speech fluency
- Effortful and slow speech
- Comprehension is intact
- Repetition is impaired like wernicke's aphasia
- The patient acknowledges with frustration their deficit
- Conduction aphasia
- Associated with lesions in the arcuate fasiculus - however this is a very simplistic view
- Speech is fluent and grammatical structure is preserved
- Comprehension is preserved
- Repetition is impaired
- Global aphasia
- The speech is non fluent and agrammatical
- The speech is also unintelligible
- Repetition is impaired
- comprehension is impaired
Myopathies
10 patterns of weakness to recognise
What is the myopathic EMG pattern?
- Proximal limb-girdle weakness
- Adult onset, painless proximal weakness
- Endocrine [steroid/ thyroid]/degenerative/toxic myopathy
- Inflammatory myositis including PM and IBM
- Neurogenic – CIPD
- Note CK may not be distinguishing feature
- Several LGMD have associated cardiomyopathy/respiratory weakness
- Arrhythmia or heart block
- Late onset pompe's disease - glycogen storage disorder
- Autosomal recessive
- Late onset limb girdle myopathy with respiratory weakness
- Dx:
- Muscle biopsy, dried blood spot test, gene testing for alpha glucosidase deficiency or acid maltase deficiency
- Adult onset, painless proximal weakness
- Distal weakness
- myotonic dystrophy
- Proximal arm, distal leg weakness (scapulo-peroneal or facio-scapulo-humeral)
- Emery-Dreifuss muscular dystrophy
- Distal arm, proximal leg weakness (characteristic of inclusion body myositis and occasional forms of muscular dystrophy)
- Ptosis and/or ophthalmoplegia: myasthenia, occulopharangeal muscular dystrophy
- Neck extensor weakness
- Bulbar weakness
- Episodic pain, weakness, myoglobuinuria with a trigger (exercise, non-exercise)
- Episodic weakness, delayed or unrelated to exercise
- Muscle stiffness, poor relaxation
- myotonic dystrophy
- stiff person syndrome
- Part of the differential diagnosis for a myopathy, especially a presentation with predominant proximal myopathy
- Duchenne MD [X-linked]
- Large truncated dystrophin protein secondary to mutations
- Progressive, symmetrical proximal limb weakness
- Childhood onset, reduced longevity
- + Contractures, scoliosis, intellectual impairment, night blindness, dilated cardiomyopathy
- 30% new mutations
- Becker MD [X-linked]
- Less truncated dystrophin protein
- Progressive, symmetrical, proximal limb weakness
- Especially quadriceps, hamstrings
- less cardiac involvement
- Milder phenotype, later onset, better survival
- 3rd commonest dystrophy
- Asymmetrical weakness
- myopathic facies
- look for winging of the scapulae
- weakness of abdominal muscles, with lower abdominal muscles weaker than upper abdominal muscles (Beevor's sign)
- bilateral weakness of dorsiflexion with foot drop
- Face especially lips, scapular, humeral, bicepts > triceps
- Hearing aids to assess for hearing (sensorineural deafness in 75% of patients)
- Fundoscopy --> retinal telangiectasia
- SPARES: deltoids, EOMs, bulbar, respiratory
- Beevor's sign - specific for facio-scapulo-humeral muscular dystrophy. Here umbilicus is displaced upwards with neck flexion in the supine position caused by asymmetrical muscular weakness
- Polymyositis and dermatomyositis
- Jo-1 [tRNA synthetase] = anti-synthetase syndrome = proximal muscle weakness + mechanics hands + ILD + systemic fever + arthritis
- Dermatomyositis specific Abs = anti-Mi-2, anti-SUMO-1
- Necrotising autoimmine myositis
- Acute onset, usually viral or drug related [esp statin]
- Classically continues even after drug withdrawal
- 19% of all inflammatory myopathies! = more common than IBM
- With statin --> worsens after drug withdrawal, if get better 4-6/52 after withdrawal probably due to statin effect
- Abs to HMG-CoAR= reductase, Signal Recognition peptide. NB statin upregulates HMG-CoA, therefore rationale for persistence of drug effect.
- Inclusion body myositis
- Epidemiology
- Most common muscle disease >50yo, previously thought steroid unresponsive polymyositis
- Sporadic
- Clinical --> lower limb proximal [with quads wasting] + weakness distally in finger flexor compartment and forearm muscles, asymmetric, dysphagia [50%], camptocormia [bending forward due to atrophy of axial muscles]
- Diagnosis --> CK elevated, EMG --> inflammatory myopathy pattern with neurogenic features, Bx à necrotising myopathy with inclusion bodies and rimmed vacuoles, note patchy involvement, can be missed
- Epidemiology
- Rx --> Steroid unresponsive, may respond to IVIG but not consistent
What is the myopathic EMG pattern?
- Small and short duration, polyphasic units [cf neurogenic = large polyphasic units with long duration]
- No spontaneous activity cf neurogenic [spontaneous fibrillation potentials]
- If myositis = +++ spontaneous activity from irritable muscle with chronic repetitive discharges
- Dive-bomber pattern if myotonic dystrophy
Friedrich's Ataxia
General inspection
What is the genetic basis of friedrich's ataxia?
What is the differential diagnosis for absent ankle jerk reflexes but up-going plantars?
- pes cavus
- hammer toe
- inversion deformity at the feet
- kyphoscoliosis
- high arched palate
- Distal wasting
- wide based gait
- positive rhomberg's
- difficulty tandem walking
- bilateral impairment in heel to shin cerebellar testing
- pyramidal pattern of weakness
- absent AJ/ KJ
- extensor plantar response
- posterior column loss (vibration, joint position sense)
- May have signs of truncal ataxia
- Distal wasting and weakness often worse in the lower limbs
- bilateral cerebellar signs, with impaired disdiadochokinesis, dysmetria andintention tremor
- scanning, slurred dysarthria with staccato speech
- hypermetric and hypometric saccades
- bilateral horizontal (fast phase) nystagmus
- jerky carotid pulse, double impulse apical beat, hypertrophic cardiomyopathy
- optic atrophy
- peripheral signs of diabetes
- 10% have sensorineural deafness
What is the genetic basis of friedrich's ataxia?
- This is an autosomal recessive ataxia with a triplet repeat expansion on chromosome 9 in the protein frataxin.
- This causes impaired mitochondrial iron regulation and subsequent iron accumulation in the mitochondria.
- This causes impaired oxidative phosphorylation
What is the differential diagnosis for absent ankle jerk reflexes but up-going plantars?
- Freidrich's ataxia
- Tabes dorsalis
- Subacute combined degeneration of the cord
- Dual pathology
- peripheral neuropathy and stroke
- peripheral neuropathy and cervical myelopathy
- cervical and lumbar spondylosis
- conus medullaris lesion
Spinocerebellar ataxias
- I will only name a few here...
- These are a diverse group of hereditary ataxia's encompassing Friedrich's ataxia
- They can be purely ataxic (SCA5, SCA6, SCA11, SCA26, SCA29, SCA30, and SCA31, known as type III SCAs), ataxia with opthalmoplegia, optic atrophy, dementia (SCA1-4)
- There are 30 types of spinocerebellar ataxia
- SCA6 is Friedrich's ataxia
- A number of the sinocerebellar ataxias are triplet repeat disorders
- SCA1: Progressive ataxia, hyper-reflexia, extensor plantar response, hypertonia. Pathologically, SCA1 is characterized by degeneration of cerebellar Purkinje cells, brainstem cranial nerve and inferior olivary nuclei, and the spinocerebellar tracts
- SCA2 distinguished from SCA1 with slow sacaddic eye movements and hyporeflexia
- SCA3 (most common in Australia): Autosomal dominant ataxia. Mutated gene on chromosome 14 with triplet repeat expansion. The mutated protein localises to the nucleus and causes nuclear inclusions and secondary degeneration. Multiple clinical features including:
- slow sacaddic eye movements
- ataxia
- lid retraction with appearance of permanent stare
- brainstem dysfunction with dysarthria, difficulty swallowing, tongue fasciculations
- mixed upper and lower motor neuron signs: tone can range from hypotonia to rigidity, reflexes can range from absent to exaggerated, and the plantar response is usually extensor
- Cognitive impairments, such as verbal and visual memory deficits, impairment of verbal fluency, and visuospatial and constructional dysfunction
- SCA5: pure cerebellar ataxia with global cerebellar atrophy, horizontal and vertical nystagmus, impaired vestibular-ocular reflex.
Cervical myelopathy
Upper limb
What is the differential diagnosis for spastic paraparesis?
- Pronator drift - sign of an upper motor neuron - may not be present
- Evidence of pseudoathetosis due to proprioceptive loss secondary to impairment of the posterior columns
- Hypotonia if the myelopathy occurs at the level (C5 - C7), hypertonia if the myelopathy occurs above the level (I was examined on a patient with C1, C2 cervical myelopathy)
- Power: lower motor neuron pattern with weakness and wasting at the level of the myelopathy, if above then perhaps a pyramidal distribution of weakness (flexors > extensors).
- Studies have shown that pyramidal distribution of weakness is an imperfect sign in distinguishing upper motor neuron from lower motor neuron lesions
- Reflexes: at the level of the myelopathy the reflexes are absent
- there may be inversion. Eg the bicep and supinator jerk may be absent however the triceps reflex is activated paradoxically.
- mid cervical pattern if there is lower motor neuron signs at C5 - C7, inverted reflexes at this level, brisk triceps jerk.
- hoffman sign
- dynamic hoffmans sign - active flexion and extension of the cervical spine brings out the positive hoffman's sign
- Co-ordination: may show evidence of pseudoathetosis
- Sensation: sensory loss in a dermatomal pattern
- usually the first limb to be involved
- Gait: bilateral scissor gait due to spasticity
- evidence of sensory ataxia
- positive rhomberg's sign
- Tone: bilateral hypertonia with increased ankle clonus
- Power: Weak in a pyramidal distribution, with extensor strength > flexor strength
- Reflexes: hyper-reflexia, crossed adductor reflex, up-going plantars
- Sensation: involvement of the posterior columns with proprioception and vibration loss
What is the differential diagnosis for spastic paraparesis?
- Friedrich's ataxia
- tabes dorsalis
- subacute degeneration of the cord
- cervical myelopathy
- multiple sclerosis
- One study showed that if all the following clinical signs were absent then this was very sensitive at ruling out cervical myelopathy. (1) gait deviation; (2) +Hoffmann’s test; (3) inverted supinator sign; (4) +Babinski test; and (5) age >45 years
Cranial Nerve III Palsy
- Clinical anatomy- lies in midbrain, at level of superior coliculus, near the cerebral aqueduct. The fascicles pas through red nucleus, substantia nigra and close to cerebral peduncle to exit brainstem. Travels very close to posterior communicating artery. Enters cavernous sinus. Anatomical ddx- midbrain lesions (consider Weber syndrome), interpeduncular cistern (transtentorial herniation), aneurysm, meningitis, mononeuritis multiplex, cavernous sinus (carotico-cavernous fistula, tumour, granuloma, thrombosis)
- Most common cause of isolated cranial nerve III palsy is ischaemic infarction causing an ischaemic mononeuropathy
- Other causes include head trauma, neoplastic, aneurysm (posterior communicating artery), idiopathic
- Clinical findings of downward and outward deviation of the affected eye, with associated ptosis due to denervation of levator palpabrae mm. Diplopia is maximal when the opposite eye is looking upwards and outwards. That is if there is a complete right third nerve palsy, then diplopia will be greatest on left upward and lateral gaze
- Isolated CNIII may be pupil sparing - which usually rules out an aneurysm as the cause. In over 95% of anaeurysmal palsies, the pupil reacts sluggishly to light or is fixed and dilated
- Anatomical correlations
- Damage as CNIII exits brainstem causes ipsilateral CNIII, ipsiateral cerebellar signs, contralateral hemitremor (due to involvement of the red nucleus) and contralateral hemiparesis due to involvement of the cerebral hemisphere
- trans-tentorial herniation and internal carotid-posterior communicating artery aneurysm causes isolated III nerve palsy
- lesions of cavenous sinus or orbit causes simultaneous injury to III, IV and VI - total opthalmoplegia, may cause small pupil due to damage to the sympathetic nerves to the iris, and to opthalmic division of V
Pupillary abnormalities
- Pupillary aperture controlled by the competing actions of two muscles - sphincter pupillae which is innervated by the parasympathetic nn and dilator pupillae which is innervated by the sympathetic nn
- Tonic pupil- adies pupil: pupil is dilated, with lack of response to light but sluggishly accomodates. When the adies pupil is associated with absent limb reflexes this is termed holmes adie pupil. Will however constrict to pilocarpine which is a parasympathetomimetic. Cause unknown.
- Argyl- Robertson pupil: small pupil irregular in shape which does not react to light but reacts to accommodation. Cause: syphilis, can occur with midbrain lesions.
- Marcus-gun pupil: this is the relative Afferent pupillary defect. Normally when shining light, the pupil constricts and there is a degree of waxing and waning - hippus. When Afferent transmission in optic nn impaired, the waning escape is more prominent.
- To detect, shine first in the affected eye (see absent direct and consensual), shine to other eye (observing direct and consensual), then shine back after 1-2 seconds- observing for pupillary dilation.
Notes on ptosis
- If third nn- complete ptosis, affected eye has divergent strabismus and is in down and out position. The pupil may be dilated or pupil may be spared.
- Senile ptosis is not associated with any other neurological abnormalities and occurs due to dehiscence of levator palpabrae superiosis from tarsal plate. Often bilateral in the elderly
- Horner's syndrome- associated with unilateral ptosis, miosis and anhydrosis. Occurs due to involvement of the superior cervical chain sympathetic nn ganglion. Can have brachial plexopathy if associated with pancoast tumour, carotid aneurysm, lateral medullary syndrome, cavernous sinus lesion.
- If associated with weakness in obicularis oculi, then ptosis may be associated with myopathy (oculopharangeal dystrophy, Kearns-Sayre syndrome, myotonic dystrophy), neuromuscular junction pathology: myaesthenia, LEMS.
Visual field defects
- The method that I use is to first ask the patient if they can see all the parts of my face
- Following this I then ask the patient to cover one eye and then go on to test each part of the vision with a red hat pin, giving clear instructions to acknowledge when the hat pin turns red
- Should also test both fields at once, to test for visual neglect
- Monocular defects tend to be pre-chiasmal
- central scotoma - characteristic of most optic nn lesions, optic nn compression (orbital lesion, lesion within optic canal, intracranial lesions) - RAPD may be pressent
- Arcuate scotoma - scharacteristic of glaucoma, with the scotoma extending from the blind spot following the course of nerve fibres
- junctional scotoma - this is immediately pre-chiasmal, in that the lesion affects the ipsilateral eye with a quantralateral defect in temporal vision as some of the nasal fibres when it decussates loops forward into the opposite optic nn
- Chiasmal defects
- Bi-temporal hemianopia: the superior quadrants are affected first if there is compression of the optic chiasm from below
- DDx: pituitary adenoma, nasopharyngeal carcinoma, sphenoid sinus, mucocele
- Bi-temporal hemianopia with the inferior quadrants affected first if the lesion originates from above the chiasm
- DDx: craniopharyngioma, third ventricular tumour
- Observe for features of tumour hyper-secretion (cushings disease, acromegaly) or hypo-secretion (panhypopituitarism)
- Bi-temporal hemianopia: the superior quadrants are affected first if there is compression of the optic chiasm from below
- Post-chiasmal defects
- congruous homonymous hemianopia: occurs due to lesions that are post chiasmal
- superior quadrantanopia occurs due to temporal lobe lesions (which receive the inferior optic radiations after synapsing with the lateral geniculate body
- inferior quadrantanopia occurs after lesions to the parietal lobe where the superior optic radiations pass through
- If dominant lobe then gerstmans syndrome with acalculia, agraphia, left right disorientation and finger agnosia
- If non-dominant lobe then sensory and visuo-spatial neglect, constructional apraxia, agraphaesthesia, astereognosis
- Test for hemiparesis or change to sensation
Binocular diplopia
- Bi-nocular diplopia occurs when the retinal image falls in different parts of each eye, and occurs due to weakness of extra-ocular muscles
- Monocular diplopia occurs as a result of refractive errors within one eye itself, and is correctable if using a pinhole
- CN VI weakness causes impaired abduction of the affected eye.
- innervates the lateral rectus muscle
- Emerges from the pons, traversing close to the medial lemniscus and emerges ventrally. Thereafter it travels upwards 1.5cm over the ventral surface of the pons and subsequently travels over the petrous portion of the temporal bone where it may become impinged
- If Left eye abducens nn palsy then diplopia greatest on attempted left lateral gaze with image occuring side by side. Covering the left eye will cause the furthest image to disappear.
- DDx: mononeuritis multiplex (diabetes, hypertension, vasculitis, toxins), raised intracranial pressure, pontine lesions, basilar infarcts, cavenous sinus pathology, involvement of the superior orbital fissure (pagets disease with hyper-osteitis), trauma
- CNIV palsy
- Innervates the superior oblique muscle which causes downward inward displacement of the eye, and intorsion
- Trochlear nn emerges from the midbrain, longest cranial nn, decussating after the nucleus and passing out at the level of the inferior colliculus.
- Defective depression in the affected eye
- Diplopia maximal when looking downwards and lateral
- Patient often tilts their head away from the affected side to minimise diplopia
- DDX: Mid-brain lesion (causes contralateral hemiparesis and hemisensory loss), cerebellar vermis masses (as it passes close to this region), raised intracranial pressure, mononeuritis multiplex, cavenous sinus pathology (thrombus, carotico-cavenous fistulae, granuloma, infection, mucocele). Most commonly associated with trauma
- CNIII palsy - see notes
- Internuclear opthalmoplegia
- Anatomy: MLF links the abducens nuclei in the pons with the contralateral CNIII nuclei
- if problem in the left medial longitudinal fasciculus then the left eye fails to adduct in participation of rightward conjugate gaze. The right eye displays nystagmus to compensate. However if the right eye is covered then the left eye is able to complete the adduction.
- Unilateral INO is commonly caused by ischaemia
- Note if there is complex opthalmoplegia that does not fit a particular cranial nerve pattern, think about structural causes such as myopathies, thyroid eye disease etc
Notes on Nystagmus
- By convention, the direction of nystagmus is defined by the fast phase.
- In peripheral nystagmus, the fast phase is directed away from the lesion and accentuates when gaze is directed towards the direction of the fast phase. Furthermore, when vision is denied, nystagmus is brought on
- In cerebellar nystagmus, the fast phase is directed towards the side of the lesion
- Physiological nystagmus
- occurs at the extremes of gaze when the eyes reach the end of the elastic properties of the connective tissue.
- Vertical nystagmus usually signifies a central lesion
- upbeat nystagmus is seen in midbrain disease - parinauds syndrome with midbrain lesion
- downbeat nystagmus is associated with lesions of the cervicomedullary junction
- dorsal medulla has input from the posterior semicircular canals, if you lose this then the eyes drift upwards and you have jerk nystagmus downwards
- lesions to the cerebellar flocculus causes downbeat nystagmus because it impairs purkinje fibres that mediate inhibition of upgaze
- DDx: arnold chiari malformation, CANVAS syndrome, lithium, hypomagnesaemia, phenytoin, carbemazepine
The wasted hand
- Innervation to the hand is provided by the median nn (first two lumbricals, opponens policis, abductor pollicis brevis and flexor pollicis brevis) and the ulnar nn (the rest of the hand intrinsic muscles - test in particular the first dorsal interossei and abductor digiti minimi)
- Ulnar nn lesions will cause (1) clawing of the hands and especially fourth and fifth finger because of involvement of the lumbricals (2) loss of sensation in an ulnar distribution
- Median nn lesions will cause (1) benediction sign with first two fingers unable to flex at the MCP and extend at IPJ due to denervation of lumbricals 1 and 2 (2) impairment in thumb opposition (3) wasting of the thenar eminence, (4) median nn pattern of sensory loss
- Disuse atrophy - but the affected mm should not be significantly weak
- Claw hand: there is hyper-extension deformity at MCP and flexion deformity at the PIP and DIPs, caused due to weakness of the lumbricals and interossei with un-opposed action of the antagonist extensor muscle grounps
- Only C8-T1 lesions (anterior horn, roots, brachial plexus) will cause the claw deformity as otherwise it would affect the extensor muscles as well
- Split hand syndrome: FDI and APB are wasted, with ADM is spared (sign of motor neuron disease), remembering that FDI and ADM are ulnar innervated mm and APB is median innervated mm. Also occurs in SCA3, autosomal dominant spinal muscular atrophy
- Horner's syndrome with C8-T1 brachial plexopathy
- Myotonic dystrophy (bilateral distal wasting of hands and feet, myopathic facies, frontal balding, cataracts, grip myotonia, percussion myotonia)
- C8-T1 cervical myelopathy - expect upper neuron findings below the lesion and weak or absent tricep jerk reflexes
- Motor neuron disease with prominent fasiculations
- syringomyelia causes ipsilateral hand weakness, areflexia and dissociated sensory loss (pain and temperature, preserved posterior columns) in a cape like distribution
- Peripheral motor neuropathy
- If there is finger/ wrist extensor involvement this usually means that there is more than just a median/ ulnar neuropathy
