Question 1
A 60-year-old woman was admitted to this hospital because of painful muscle spasms, weakness, and hyperreflexia.The patient had been well until approximately 4 months before admission, when stiffness and cramping developed in both legs, associated with pain in the back. Two weeks before admission, muscle cramps in the legs progressively worsened. The cramps were more severe proximally, occurred after movement or pressure on the legs, and awakened the patient from sleep. In the days before admission, spasms and stiffness led to multiple falls and required use of a walker. Symptoms were exacerbated by emotions and sensory stimuli. Investigations included MRI brain which was unremarkable, whilst MRI spine showed degenerative changes only. Serum testing was significant for anti-GAD antibodies. Her management included that of intravenous immunoglobulin. The most likely explanation for this presentation is which of the following?
(A) Stiff person syndrome
(B) myotonic dystrophy
(C) limb girdle muscular dystrophy
(D) spinocerebellar ataxia 2
(E) Freidrich's ataxia
A: The progressive stiffness, spasms and hyperreflexia with positive anti-GAD antibodies is a clue to the diagnosis. Chronic pain, rigidity and lumbar hyperlordosis is also another manifestation. The lumbar and abdominal muscles engage in constant contractions. Stress, cold weather, and infections lead to an increase in symptoms, and sleep decreases them. European Journal of Clinical Investigation 40 (11): 1018–25.
Question 2
A 68 year old male with progressive weakness is referred to the neurology clinic for further investigation. He notes a tendency to fall forward, difficulty drinking cups of coffee and difficulty getting up from a seated position. Examination findings show weak finger flexors and quadriceps wasting. There is eczema on both his hands. He has no other rashes. He uses 10mg pravastatin, which was prescribed for uncomplicated dyslipidaemia. He has a positive ANA at 1:40 but no detectable ENA or ANCA. His CK is 3000. EMG shows spontaneous discharge and myopathic recordings with small polyphasic spikes. A muscle biopsy is organised and a decision is made to begin treatment whilst waiting for the crucial results.
What treatment would you institute?
(A) cyclophosphamide
(B) methylprednisolone
(C) azathioprine
(D) IVIG
(E) mycophenylate
D: The most likely diagnosis is inclusion body myositis given the patients age. This is characterised by proximal myopathy with characteristic distal finger flexor involvement. It is sporadic. A myositis EMG is seen with spontaneous potentials and low amplitude polyphasic spikes. A biopsy would reveal rimmed vacuoles and inclusion bodies. It is a difficult condition to treat and is characteristically steroid non-responsive. Anti-CN1a may be positive. N Engl J Med 2015; 372:1734-1747
Question 3
A 62 year old female presents to the clinic with acute onset limb girdle pattern myopathy. She has been taking simvastatin 20mg daily after a CVA 8 months ago. An initial diagnosis of statin induced myopathy is made, however after abrupt withdrawal of the drug, the patient is found to have progressive symptoms. Antibodies for HMG-CoA reductase and signal recognition peptide (anti-SRP) are present.
What is the most likely diagnosis?
(A) autoimmune necrotising myositis
(B) polymyositis
(C) myotonic dystrophy
(D) atypical variant statin induced myopathy
(E) beckers muscular dystrophy
A: This is classic of ANM. It is usually acute or subacute onset after initiation of the drug, and characteristically worsens after removal, however 4 - 6 weeks later the pattern should get better. Implicated antibodies include anti-HMGR and anti-SRP. EMG would show active myopathic units. Polymyositis would typically be associated with a stem involving the anti-sdnthetase syndrome (Jo1 positive, ILD, mechanics hands and proximal myopathy), myotonic dystrophy stem would include a hereditary anticipation phenomenon with frontal balding, intellectual disability and cardiomyopathy. The dystrinopathies would involve a stem that included a pedigree showing X-linked inheritance with a cardiomyopathy. N Engl J Med 2015; 372:1734-1747
Question 4
A 36 year old male has a prolonged ICU course for sepsis complicated by ARDS. He was ventilator dependent for 50 days, required renal replacement therapy and at one time had a profound ischaemic hepatitis. He is currently on a tracheostomy and on the lowest cuff size. He is referred to neurology as he suffers from weakness. This weakness is characterised by preserved sensation and a proximal pattern to the injury. Nerve conduction studies show increased distal latencies and reduced CMAP potentials. Her creatine kinase is not elevated.
What is the most likely diagnosis?
(A) Critical illness polyneuropathy
(B) Critical illness myopathy
(C) Steroid induced myopathy
(D) Cocksaxie B myopathy
(E) Guillan barre' syndrome
B: The prolonged ICU course in the stem indicates that the patient has either critical illness polyneuropathy or critical illness myopathy. The history and examination do not give much distinguishing features. The NCS is helpful. If there is increased distal latencies and reduced CMAP potentials as there is in this case then the patient has a myopathy. If the CMAP potential is decreased, but distal latencies are presented then the answer would be a polyneuropathy. The reason is that a polyneuropathy is axonal and not demyelinating. It can involve the anterior horn cells. A myopathy biopsy would show necrosis and selective reduction of thick filaments owing to the degradation of myosin protein. N Engl J Med 2014; 370:1626-1635
Question 5
A 68 year old male presents with bilateral ptosis, which he says is worse after the end of the day. He also tells you that he is feeling difficulty in breathing, and is exhausted after climbing a flight of stairs. On examination he has bilateral ptosis. His autoantibodies show AchR (neg), MUSK (pos), PRP4 (neg). What is TRUE about the treatment of this condition
(A) He has MUSK + myasthenia gravis, and requires prednisolone in conjunction with acetylcholinesterase inhibitors
(B) He has MUSK + myasthenia gravis, and requires Plasma exchange in conjunction with acetylcholinesterase inhibitors
(C) He has MUSK + myasthenia gravis, and requires IVIG in conjunction with acetylcholinesterase inhibitors
(D) He has MUSK + myasthenia gravis, and requires Cyclophosphamide in conjunction with acetylcholinesterase inhibitors
(E) He has MUSK + myasthenia gravis, and requires further repetitive stimulation studies to document a decrease in action potential
B: For MUSK, immunosuppression tends not to work so well, and plasma exchange is favoured over IVIG. Prednisone should not be used in the first two weeks of treatment as it may exacerbate respiratory depression. MUSK + phenotype tends to favour generalisable MG over pure ocular MG. Pure ocular MG carries with it the best prognosis. Bulbar and respiratory weakness, however, is common. Smooth muscle and cardiac muscle are not involved in myasthenia gravis. NCS and EMG are not helpful in the diagnosis of MG, however repetitive stimulation studies may help confirm a diagnosis. Note that in MUSK + myasthenia graves, the tension test (adding an acetylcholinesterase inhibitor) usually is unyielding. RPA course 2015
Question 6
The following image was taken from a patient exhibiting signs of a disease. What is the most likely diagnosis (Courtesy of: doi:10.1016/S0140-6736(10)61156-7 Lancet, Volume 377, Issue 9769, 12–18 March 2011, Pages 942–955)
A 60-year-old woman was admitted to this hospital because of painful muscle spasms, weakness, and hyperreflexia.The patient had been well until approximately 4 months before admission, when stiffness and cramping developed in both legs, associated with pain in the back. Two weeks before admission, muscle cramps in the legs progressively worsened. The cramps were more severe proximally, occurred after movement or pressure on the legs, and awakened the patient from sleep. In the days before admission, spasms and stiffness led to multiple falls and required use of a walker. Symptoms were exacerbated by emotions and sensory stimuli. Investigations included MRI brain which was unremarkable, whilst MRI spine showed degenerative changes only. Serum testing was significant for anti-GAD antibodies. Her management included that of intravenous immunoglobulin. The most likely explanation for this presentation is which of the following?
(A) Stiff person syndrome
(B) myotonic dystrophy
(C) limb girdle muscular dystrophy
(D) spinocerebellar ataxia 2
(E) Freidrich's ataxia
A: The progressive stiffness, spasms and hyperreflexia with positive anti-GAD antibodies is a clue to the diagnosis. Chronic pain, rigidity and lumbar hyperlordosis is also another manifestation. The lumbar and abdominal muscles engage in constant contractions. Stress, cold weather, and infections lead to an increase in symptoms, and sleep decreases them. European Journal of Clinical Investigation 40 (11): 1018–25.
Question 2
A 68 year old male with progressive weakness is referred to the neurology clinic for further investigation. He notes a tendency to fall forward, difficulty drinking cups of coffee and difficulty getting up from a seated position. Examination findings show weak finger flexors and quadriceps wasting. There is eczema on both his hands. He has no other rashes. He uses 10mg pravastatin, which was prescribed for uncomplicated dyslipidaemia. He has a positive ANA at 1:40 but no detectable ENA or ANCA. His CK is 3000. EMG shows spontaneous discharge and myopathic recordings with small polyphasic spikes. A muscle biopsy is organised and a decision is made to begin treatment whilst waiting for the crucial results.
What treatment would you institute?
(A) cyclophosphamide
(B) methylprednisolone
(C) azathioprine
(D) IVIG
(E) mycophenylate
D: The most likely diagnosis is inclusion body myositis given the patients age. This is characterised by proximal myopathy with characteristic distal finger flexor involvement. It is sporadic. A myositis EMG is seen with spontaneous potentials and low amplitude polyphasic spikes. A biopsy would reveal rimmed vacuoles and inclusion bodies. It is a difficult condition to treat and is characteristically steroid non-responsive. Anti-CN1a may be positive. N Engl J Med 2015; 372:1734-1747
Question 3
A 62 year old female presents to the clinic with acute onset limb girdle pattern myopathy. She has been taking simvastatin 20mg daily after a CVA 8 months ago. An initial diagnosis of statin induced myopathy is made, however after abrupt withdrawal of the drug, the patient is found to have progressive symptoms. Antibodies for HMG-CoA reductase and signal recognition peptide (anti-SRP) are present.
What is the most likely diagnosis?
(A) autoimmune necrotising myositis
(B) polymyositis
(C) myotonic dystrophy
(D) atypical variant statin induced myopathy
(E) beckers muscular dystrophy
A: This is classic of ANM. It is usually acute or subacute onset after initiation of the drug, and characteristically worsens after removal, however 4 - 6 weeks later the pattern should get better. Implicated antibodies include anti-HMGR and anti-SRP. EMG would show active myopathic units. Polymyositis would typically be associated with a stem involving the anti-sdnthetase syndrome (Jo1 positive, ILD, mechanics hands and proximal myopathy), myotonic dystrophy stem would include a hereditary anticipation phenomenon with frontal balding, intellectual disability and cardiomyopathy. The dystrinopathies would involve a stem that included a pedigree showing X-linked inheritance with a cardiomyopathy. N Engl J Med 2015; 372:1734-1747
Question 4
A 36 year old male has a prolonged ICU course for sepsis complicated by ARDS. He was ventilator dependent for 50 days, required renal replacement therapy and at one time had a profound ischaemic hepatitis. He is currently on a tracheostomy and on the lowest cuff size. He is referred to neurology as he suffers from weakness. This weakness is characterised by preserved sensation and a proximal pattern to the injury. Nerve conduction studies show increased distal latencies and reduced CMAP potentials. Her creatine kinase is not elevated.
What is the most likely diagnosis?
(A) Critical illness polyneuropathy
(B) Critical illness myopathy
(C) Steroid induced myopathy
(D) Cocksaxie B myopathy
(E) Guillan barre' syndrome
B: The prolonged ICU course in the stem indicates that the patient has either critical illness polyneuropathy or critical illness myopathy. The history and examination do not give much distinguishing features. The NCS is helpful. If there is increased distal latencies and reduced CMAP potentials as there is in this case then the patient has a myopathy. If the CMAP potential is decreased, but distal latencies are presented then the answer would be a polyneuropathy. The reason is that a polyneuropathy is axonal and not demyelinating. It can involve the anterior horn cells. A myopathy biopsy would show necrosis and selective reduction of thick filaments owing to the degradation of myosin protein. N Engl J Med 2014; 370:1626-1635
Question 5
A 68 year old male presents with bilateral ptosis, which he says is worse after the end of the day. He also tells you that he is feeling difficulty in breathing, and is exhausted after climbing a flight of stairs. On examination he has bilateral ptosis. His autoantibodies show AchR (neg), MUSK (pos), PRP4 (neg). What is TRUE about the treatment of this condition
(A) He has MUSK + myasthenia gravis, and requires prednisolone in conjunction with acetylcholinesterase inhibitors
(B) He has MUSK + myasthenia gravis, and requires Plasma exchange in conjunction with acetylcholinesterase inhibitors
(C) He has MUSK + myasthenia gravis, and requires IVIG in conjunction with acetylcholinesterase inhibitors
(D) He has MUSK + myasthenia gravis, and requires Cyclophosphamide in conjunction with acetylcholinesterase inhibitors
(E) He has MUSK + myasthenia gravis, and requires further repetitive stimulation studies to document a decrease in action potential
B: For MUSK, immunosuppression tends not to work so well, and plasma exchange is favoured over IVIG. Prednisone should not be used in the first two weeks of treatment as it may exacerbate respiratory depression. MUSK + phenotype tends to favour generalisable MG over pure ocular MG. Pure ocular MG carries with it the best prognosis. Bulbar and respiratory weakness, however, is common. Smooth muscle and cardiac muscle are not involved in myasthenia gravis. NCS and EMG are not helpful in the diagnosis of MG, however repetitive stimulation studies may help confirm a diagnosis. Note that in MUSK + myasthenia graves, the tension test (adding an acetylcholinesterase inhibitor) usually is unyielding. RPA course 2015
Question 6
The following image was taken from a patient exhibiting signs of a disease. What is the most likely diagnosis (Courtesy of: doi:10.1016/S0140-6736(10)61156-7 Lancet, Volume 377, Issue 9769, 12–18 March 2011, Pages 942–955)
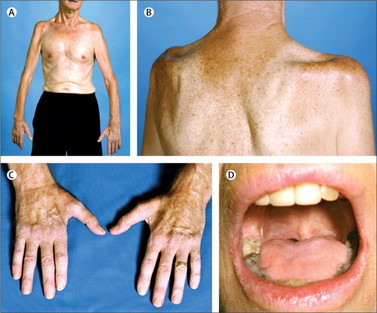
(A) Amyotrophic Lateral Sclerosis
(B) Myotonic Dystrophy
(C) AIDP
(D) CIDP
(E) Facial-Scapular-Humeral Muscular Dystrophy
A: See the common sign of 'split hand syndrome', toungue wasting, wasting of infraspinatus, supraspinatus and prominence of the glenohumeral joint.
(B) Myotonic Dystrophy
(C) AIDP
(D) CIDP
(E) Facial-Scapular-Humeral Muscular Dystrophy
A: See the common sign of 'split hand syndrome', toungue wasting, wasting of infraspinatus, supraspinatus and prominence of the glenohumeral joint.
