Question 1
What is TRUE about the role of hepcidin in iron homeostasis
(A) Hepcidin is not implicated in the pathogenesis of hereditary haemochromatosis
(B) Hepcidin is synthesied by renal tubular cells in response to hypoxia
(C) Hepcidin decreases the synthesis of transferrin decreasing the transport of iron
(D) Hepcidin decreases the expression of the divalent metal cation transporter in enterocytes
(E) Hepcidin is an acute phase reactant that binds to ferroportin and causes internalisation and ultimately degradation of the transporter
E: Hepcidin as an acute phase reactant. Cytokines such as IL-6 can acutely cause an increase in hepcidin levels. None of the other mechanisms are correct. Increased hepcidin levels in chronic inflamamtory anaemia are tyhought to be one of the primary mechanisms. Camaschella, C. Iron-Deficiency Anemia (2015) NEJM; 372:1832-1843
Question 2
What is TRUE about the mechanism of hereditary Iron Refractory Iron Deficiency Anaemia (IRIDA)
(A) Mutations in TMPRSS6, encoding matriptase-2, causes loss of function mutations. Matriptase 2 is involved in inhibiting signalling pathways that activate hepcidin.
(B) Gain of function mutations in SFX65-box 3 domain causes decreased hepcidin degradation
(C) subtherepeutic iron infusions can overcome the relative resistance and should be prescribed
(D) Mutations in the divalent metal transporter accound for this condition
(E) Hypoxia inducible factor alpha is at supraphysiological levels in this condition
A: All other options I made up! IRIDa is diagnosed on the basis of a failure of haematological response to iron after 4 – 6 weeks of Fe therapy (<1g/dL increment)
Question 3
What is FALSE regarding the association between helicobacter pylori and iron deficiency anaemia
(A) H. pylori may cause iron deficiency anaemia through gastric erosions
(B) H. pylori may cause iron deficiency anaemia through competing for Fe
(C) H. pylori may reduce the bioavailability of vitamin C
(D) H. pylori may cause MALT lymphoma
(E) H. pylori may decrease the synthesis of intrinsic factor
E: All the others may be associated with Fe deficiency anaemia. Option E is stupid. In patients with Fe- refractory Fe-deficiency anaemia, H. pylori testing should be offered. Camaschella, C. Iron-Deficiency Anemia (2015) NEJM; 372:1832-1843
Question 4
A 46-year-old woman was admitted with a 7-day history of malaise, lethargy, bloody diarrhoea, haematuria and acute dyspnoea. She is a cattle worker on a farm. She is pyrexial and appears jaundice. There is evidence of pulmonary oedema with a fine petechial rash over her face. Her blood pressure is elevated at 190/90. There is no neurological abnormality identified. Urinalysis shows red cell casts with blood and protein. The following investigations are listed below:
haemoglobin 62 g/L (115–165)
platelet count 43 × 109/L (150–400)
prothrombin time18.0 s (11.5–15.5)
activated partial thromboplastin time 56 s (30–40)
serum sodium 140 mmol/L (137–144)
serum urea 37.0 mmol/L (2.5–7.0)
serum creatinine 480 µmol/L (60–110)
serum total bilirubin 93 µmol/L (1–22)
serum lactate dehydrogenase 2630 U/L (10–250)
Blood film: shistocytes, helmet cells and thrombocytopenia
What is the most likely underlying diagnosis?
(A) DIC
(B) TTP
(C) atypical HUS
(D) HUS
(E) PNH
D: This patient has classical features of haemolytic ureic syndrome. Note that shiga toxin producing E. coli are common commensals in cattle. The strain O157:H7 is the most common shiga toxin producing E. Coli strain. Mechanism of cell damage is through binding to Gb3, also known as CD77 with resulting damage to endothelial cells and renal mesangial and epithelial cells. Apoptosis, endocytosis, retrograde transport, cytosolic translocation and subsequent ribosomal activation occurs upon binding of toxin to Gb3 (CD77). Concomitant cell activation occurs with up-regulation of pro-inflammatory and prothrombotic cytokines. There is also increased secretion of von Willebrand factor. N Engl J Med 2014; 371:654-666
Question 5
What is associated with acquired TTP?
(A) Black race
(B) Male sex
(C) Age greater than 50
(D) Age younger than 10
(E) Scleroderma
A: Black race, female sex and age between 10 - 50 are associated with acquired TTP. Pediatr Blood Cancer 2013;60:1676-1682
Question 6
Which of the following statements about the clinical manifestation of TTP is FALSE?
(A) Neurological abnormalities occur commonly
(B) A microangiopathic haemolytic anaemia is found
(C) Thrombocytopenia may occur
(D) Acute kidney injury occurs commonly
(E) ADAMTS13 factor levels are low
D: Compared to other microangiopathic haemolytic anaemias, TTP rarely involves acute renal failure. N Engl J Med 2014; 371:654-666
Question 7
A 36 year old black female presents with sudden onset left hemiparesis, a purpuric rash and confusion. Investigations reveal a microangiopathic haemolytic anaemia, thrombocytopenia without evidence of coagulopathy or renal failure. ADAMTS13 activity and levels are decreased. There is an absence of ADAMTS13 autoantibody levels.
What is TRUE regarding the correct interpretation of the diagnostic tests?
(A) TTP likely, hereditary, ADAMTS13 genetic mutations are required
(B) atypical HUS likely
(C) HUS likely
(D) TTP confirmed, acquired, ADAMTS13 genetic mutations are not required
(E) drug induced TTP likely, ADAMTS13 genetic mutations are not required
A: The decrease in levels of ADAMTS13 combined with the lack of an auto-antibody that would decrease the function of the vWF cleaving protease would lead to a very likely diagnosis of hereditary TTP. Either the patient is homozygous or compound heterozygote. Note that management is with plasma exchange, as this repletes the ADAMTS13. N Engl J Med 2014; 371:654-666, Nature 2001;413:488-494
Question 8
What is the function of ADAMTS13?
(A) Prevents di-sulfide bridging of vWF multimers
(B) Cleaves A2 domain of large vWF multimers
(C) Cleaves B6 domain of large vWF multimers
(D) Decreases synthesis of vWF multimers by transcriptional regulation
(E) allosterically inhibits the function of vWF
B: ADAMTS13 acts via help from calcium to cleave high molecular weight multimers of vWF through the A2 domain. Blood 1996;87:4235-4244
Question 9
Which of the following factors is NOT implicated in the pathophysiology of complement mediated thrombotic microangiopathy?
(A) Deficiency in Factor H preventing dissociation of C3bBb, the alternative pathway C3 convertase
(B) Deficiency in Factor I, causing degradation of C3b into iC3b
(C) Properdin deficiency
(D) Increased susceptibility to form C3b
(E) Formation of C5b --> C6 --> C9 MAC attack complex and cell lysis with pro-thrombotic inflammation
C: Properdin is a compound that stabilises C3bBb, the alternative pathway C3 convertase. Deficiency of properdin is in fact an immunodeficiency state with susceptibility to meningococcal sepsis. Eculizumab or plasma infusions/ plasma exchange may be used to treat this condition. N Engl J Med 2013;368:2169-2181, N Engl J Med 2014
Question 10
The following image shows the metabolic pathway for the formation of succinyl-CoA and methionine from cobalamin and methyl-tetrahydrofolate. Mutation in which component is associated with metabolism associated thrombotic microangiopathic anaemia?
What is TRUE about the role of hepcidin in iron homeostasis
(A) Hepcidin is not implicated in the pathogenesis of hereditary haemochromatosis
(B) Hepcidin is synthesied by renal tubular cells in response to hypoxia
(C) Hepcidin decreases the synthesis of transferrin decreasing the transport of iron
(D) Hepcidin decreases the expression of the divalent metal cation transporter in enterocytes
(E) Hepcidin is an acute phase reactant that binds to ferroportin and causes internalisation and ultimately degradation of the transporter
E: Hepcidin as an acute phase reactant. Cytokines such as IL-6 can acutely cause an increase in hepcidin levels. None of the other mechanisms are correct. Increased hepcidin levels in chronic inflamamtory anaemia are tyhought to be one of the primary mechanisms. Camaschella, C. Iron-Deficiency Anemia (2015) NEJM; 372:1832-1843
Question 2
What is TRUE about the mechanism of hereditary Iron Refractory Iron Deficiency Anaemia (IRIDA)
(A) Mutations in TMPRSS6, encoding matriptase-2, causes loss of function mutations. Matriptase 2 is involved in inhibiting signalling pathways that activate hepcidin.
(B) Gain of function mutations in SFX65-box 3 domain causes decreased hepcidin degradation
(C) subtherepeutic iron infusions can overcome the relative resistance and should be prescribed
(D) Mutations in the divalent metal transporter accound for this condition
(E) Hypoxia inducible factor alpha is at supraphysiological levels in this condition
A: All other options I made up! IRIDa is diagnosed on the basis of a failure of haematological response to iron after 4 – 6 weeks of Fe therapy (<1g/dL increment)
Question 3
What is FALSE regarding the association between helicobacter pylori and iron deficiency anaemia
(A) H. pylori may cause iron deficiency anaemia through gastric erosions
(B) H. pylori may cause iron deficiency anaemia through competing for Fe
(C) H. pylori may reduce the bioavailability of vitamin C
(D) H. pylori may cause MALT lymphoma
(E) H. pylori may decrease the synthesis of intrinsic factor
E: All the others may be associated with Fe deficiency anaemia. Option E is stupid. In patients with Fe- refractory Fe-deficiency anaemia, H. pylori testing should be offered. Camaschella, C. Iron-Deficiency Anemia (2015) NEJM; 372:1832-1843
Question 4
A 46-year-old woman was admitted with a 7-day history of malaise, lethargy, bloody diarrhoea, haematuria and acute dyspnoea. She is a cattle worker on a farm. She is pyrexial and appears jaundice. There is evidence of pulmonary oedema with a fine petechial rash over her face. Her blood pressure is elevated at 190/90. There is no neurological abnormality identified. Urinalysis shows red cell casts with blood and protein. The following investigations are listed below:
haemoglobin 62 g/L (115–165)
platelet count 43 × 109/L (150–400)
prothrombin time18.0 s (11.5–15.5)
activated partial thromboplastin time 56 s (30–40)
serum sodium 140 mmol/L (137–144)
serum urea 37.0 mmol/L (2.5–7.0)
serum creatinine 480 µmol/L (60–110)
serum total bilirubin 93 µmol/L (1–22)
serum lactate dehydrogenase 2630 U/L (10–250)
Blood film: shistocytes, helmet cells and thrombocytopenia
What is the most likely underlying diagnosis?
(A) DIC
(B) TTP
(C) atypical HUS
(D) HUS
(E) PNH
D: This patient has classical features of haemolytic ureic syndrome. Note that shiga toxin producing E. coli are common commensals in cattle. The strain O157:H7 is the most common shiga toxin producing E. Coli strain. Mechanism of cell damage is through binding to Gb3, also known as CD77 with resulting damage to endothelial cells and renal mesangial and epithelial cells. Apoptosis, endocytosis, retrograde transport, cytosolic translocation and subsequent ribosomal activation occurs upon binding of toxin to Gb3 (CD77). Concomitant cell activation occurs with up-regulation of pro-inflammatory and prothrombotic cytokines. There is also increased secretion of von Willebrand factor. N Engl J Med 2014; 371:654-666
Question 5
What is associated with acquired TTP?
(A) Black race
(B) Male sex
(C) Age greater than 50
(D) Age younger than 10
(E) Scleroderma
A: Black race, female sex and age between 10 - 50 are associated with acquired TTP. Pediatr Blood Cancer 2013;60:1676-1682
Question 6
Which of the following statements about the clinical manifestation of TTP is FALSE?
(A) Neurological abnormalities occur commonly
(B) A microangiopathic haemolytic anaemia is found
(C) Thrombocytopenia may occur
(D) Acute kidney injury occurs commonly
(E) ADAMTS13 factor levels are low
D: Compared to other microangiopathic haemolytic anaemias, TTP rarely involves acute renal failure. N Engl J Med 2014; 371:654-666
Question 7
A 36 year old black female presents with sudden onset left hemiparesis, a purpuric rash and confusion. Investigations reveal a microangiopathic haemolytic anaemia, thrombocytopenia without evidence of coagulopathy or renal failure. ADAMTS13 activity and levels are decreased. There is an absence of ADAMTS13 autoantibody levels.
What is TRUE regarding the correct interpretation of the diagnostic tests?
(A) TTP likely, hereditary, ADAMTS13 genetic mutations are required
(B) atypical HUS likely
(C) HUS likely
(D) TTP confirmed, acquired, ADAMTS13 genetic mutations are not required
(E) drug induced TTP likely, ADAMTS13 genetic mutations are not required
A: The decrease in levels of ADAMTS13 combined with the lack of an auto-antibody that would decrease the function of the vWF cleaving protease would lead to a very likely diagnosis of hereditary TTP. Either the patient is homozygous or compound heterozygote. Note that management is with plasma exchange, as this repletes the ADAMTS13. N Engl J Med 2014; 371:654-666, Nature 2001;413:488-494
Question 8
What is the function of ADAMTS13?
(A) Prevents di-sulfide bridging of vWF multimers
(B) Cleaves A2 domain of large vWF multimers
(C) Cleaves B6 domain of large vWF multimers
(D) Decreases synthesis of vWF multimers by transcriptional regulation
(E) allosterically inhibits the function of vWF
B: ADAMTS13 acts via help from calcium to cleave high molecular weight multimers of vWF through the A2 domain. Blood 1996;87:4235-4244
Question 9
Which of the following factors is NOT implicated in the pathophysiology of complement mediated thrombotic microangiopathy?
(A) Deficiency in Factor H preventing dissociation of C3bBb, the alternative pathway C3 convertase
(B) Deficiency in Factor I, causing degradation of C3b into iC3b
(C) Properdin deficiency
(D) Increased susceptibility to form C3b
(E) Formation of C5b --> C6 --> C9 MAC attack complex and cell lysis with pro-thrombotic inflammation
C: Properdin is a compound that stabilises C3bBb, the alternative pathway C3 convertase. Deficiency of properdin is in fact an immunodeficiency state with susceptibility to meningococcal sepsis. Eculizumab or plasma infusions/ plasma exchange may be used to treat this condition. N Engl J Med 2013;368:2169-2181, N Engl J Med 2014
Question 10
The following image shows the metabolic pathway for the formation of succinyl-CoA and methionine from cobalamin and methyl-tetrahydrofolate. Mutation in which component is associated with metabolism associated thrombotic microangiopathic anaemia?
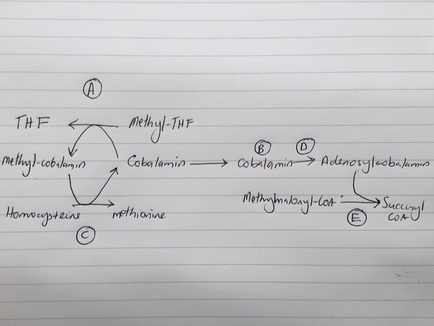
B: MMACHC deficiency leads to a functional decrease in cobalamin. This subsequently causes decreased methylation of cobalamin and hence decreased methionine synthesis with increased homocysteine levels. Methionine is an amino acid, and hyperhomocysteinaemia leads to a pro-thrombotic state. Furthermore, there is decreased synthesis of succinyl CoA leading to increased methylmalonyl CoA with methylmalonic academia. Semin Thromb Hemost 2000;26:243-254
Question 11
What is the most common cause of vitamin B12 deficiency?
(A) Pernicious anaemia
(B) H. pylori
(C) short gut syndrome
(D) Vegan diet
(E) coeliacs disease
A: "The prevalence of pernicious anemia ranges from 50 to 4000 cases per 100,000 persons, depending on the diagnostic criteria. All age groups are affected, but the median age range in large series is 70 to 80 years" Clin Rev Allergy Immunol 2012;42:269-278, N Engl J Med 2013; 368:149-160
Question 12
Which of the following common laboratory markers may help distinguish B12 deficiency from folate deficiency?
(A) elevated homocysteine
(B) elevated methylmalonic acid
(C) decreased intrinsic factor levels
(D) Red cell scan
(E) Anti-nucleolar immunofluorescence and chromatography
B: homocysteine levels are elevated in folate and B12 deficiency, whilst methylmalonic acid levels are elevated in B12 deficiency. The reason is that adenylated-B12 is a co-enzyme in the conversion of methylmalonyl CoA into succinyl CoA. The other important role of B12 is the synthesis of methionine from homocysteine. Methionine is simply the methylated version of homocysteine, and the methylation reaction requires both vitamin B12 (cobalamin) and methyl-tetrahydrofolate. Methionine synthase catalyses the transfer of the methyl group from methyl-tetrahydrofolate to B12, forming methyl-B12. Subsequently the methyl group is transferred back from methyl-B12 --> B12, generating homocysteine --> methionine. The tetrahydrofolate can be used for pyrimidine and purine synthesis. Clinical hematology. Philadelphia: Mosby, 2006:242-51. J Inherit Metab Dis. 2011 Feb; 34(1): 75–81.
Question 13
What is FALSE regarding the neurological manifestations of vitamin B12 deficiency?
(A) Cervical dorsal columns may be affected
(B) Cranial and peripheral nerves may be affected
(C) Cerebral white matter lesions may be evident
(D) Cerebral grey matter lesions may be evident
(E) Neurological manifestations may occur despite the presence of anaemia
D: Neurological manifestations may be grouped into what is known as subacute combined degeneration. Grey matter is not a feature. The most common neurologic symptoms are symmetric paresthesias or numbness and gait problems. Neurological manifestations may occur without anaemia and can be reversed with treatment. In fact one study showed that neurological manifestations are inversely correlated to the degree of megaloblastic anaemia. IM B12 is equivalent to high dose oral B12 and thus this is now the preferred option. This is because 0.5 - 4% of B12 is absorbed passively, and therefore high oral doses (1000ug) can still deliver the same requirement. Blood1998;92:1191-1198, Acta Med Scand1968;184:247-258, Clin Ther 2003;25:3124-3134
Question 14
You are reviewing the blood film of a patient with microcytic anaemia and subsequently perform a haemoglobin electrophoresis. The results show elevation in HbH, however HbA2 and HbF are reduced. Which of the following conditions is this patient likely to have
(A) Beta thalassemia minor
(B) Beta thalassemia major
(C) Alpha thalassemia trait
(D) Hydrops Fetalis
(E) Alpha thalassemia - HbH disease
E: The name implies it. The alpa haemaglobin gene contains two pairs on chromosome 16, accounting for 4 copies. Trait 1 and trait 2 results in one or two mutations. Trait 2 may be cis or trans. Mediterraneans and South East Asians tend to have the cis form, whilst trans is found more in Africa. HbH disease results from three mutations, with only one functional alpha gene. As a result, beta globins precipitate as beta 4. This leads to decreased production, increased haemolysis, target cells and HbH inclusions on peripheral film. Patients become transfusion dependent, demonstrate extramedullary haematopoiesis and may demonstrate iron overload. Hydrops fetalis results from mutations to all four alpha globulin genes and causes barts Hb = gamma 4. Beta thalassemia results in increased HbA2 concentrations and HbF concentrations.N Engl J Med 2014; 371:1908-1916
Question 15
Which condition results in the lowest level of hepcidin?
(A) Thalassemia intermedia
(B) Thalassemia major
(C) Thalassemia minor
(D) Anaemia of chronic disease
(E) Anaemia of renal failure
A: In thalassemia intermedia, patients are not transfused thus there is a high erythropoietic drive secondary to haemolysis and innefective erythropoiesis. The increased EPO results in decreased hepcidin. Thal major leads to secondary iron accumulation secondary to transfusional dependence. This increases iron levels, increases iron sensing and decreases hepatic synthesis of hepcidin. Hepcidin is increased in anaemia of chronic disease and anaemia of renal failure. This causes downregulation and internalisation of ferroportin therefore decreased Fe absorption and release of Fe from body stores. Origa et al, haematologica 2007
Question 16
Patients with beta-thalassaemia are at risk of infection. Which of the following infections is NOT associated with this condition
(A) Haemophilus
(B) Pneumococcal
(C) Yersinia
(D) Staphylococcal
(E) Meningococcal
D: Encapsulated organisms and yersinia (because it favours iron laden tissues) is particularly associated with beta-thalassaemia. RPA Course 2015
Question 11
What is the most common cause of vitamin B12 deficiency?
(A) Pernicious anaemia
(B) H. pylori
(C) short gut syndrome
(D) Vegan diet
(E) coeliacs disease
A: "The prevalence of pernicious anemia ranges from 50 to 4000 cases per 100,000 persons, depending on the diagnostic criteria. All age groups are affected, but the median age range in large series is 70 to 80 years" Clin Rev Allergy Immunol 2012;42:269-278, N Engl J Med 2013; 368:149-160
Question 12
Which of the following common laboratory markers may help distinguish B12 deficiency from folate deficiency?
(A) elevated homocysteine
(B) elevated methylmalonic acid
(C) decreased intrinsic factor levels
(D) Red cell scan
(E) Anti-nucleolar immunofluorescence and chromatography
B: homocysteine levels are elevated in folate and B12 deficiency, whilst methylmalonic acid levels are elevated in B12 deficiency. The reason is that adenylated-B12 is a co-enzyme in the conversion of methylmalonyl CoA into succinyl CoA. The other important role of B12 is the synthesis of methionine from homocysteine. Methionine is simply the methylated version of homocysteine, and the methylation reaction requires both vitamin B12 (cobalamin) and methyl-tetrahydrofolate. Methionine synthase catalyses the transfer of the methyl group from methyl-tetrahydrofolate to B12, forming methyl-B12. Subsequently the methyl group is transferred back from methyl-B12 --> B12, generating homocysteine --> methionine. The tetrahydrofolate can be used for pyrimidine and purine synthesis. Clinical hematology. Philadelphia: Mosby, 2006:242-51. J Inherit Metab Dis. 2011 Feb; 34(1): 75–81.
Question 13
What is FALSE regarding the neurological manifestations of vitamin B12 deficiency?
(A) Cervical dorsal columns may be affected
(B) Cranial and peripheral nerves may be affected
(C) Cerebral white matter lesions may be evident
(D) Cerebral grey matter lesions may be evident
(E) Neurological manifestations may occur despite the presence of anaemia
D: Neurological manifestations may be grouped into what is known as subacute combined degeneration. Grey matter is not a feature. The most common neurologic symptoms are symmetric paresthesias or numbness and gait problems. Neurological manifestations may occur without anaemia and can be reversed with treatment. In fact one study showed that neurological manifestations are inversely correlated to the degree of megaloblastic anaemia. IM B12 is equivalent to high dose oral B12 and thus this is now the preferred option. This is because 0.5 - 4% of B12 is absorbed passively, and therefore high oral doses (1000ug) can still deliver the same requirement. Blood1998;92:1191-1198, Acta Med Scand1968;184:247-258, Clin Ther 2003;25:3124-3134
Question 14
You are reviewing the blood film of a patient with microcytic anaemia and subsequently perform a haemoglobin electrophoresis. The results show elevation in HbH, however HbA2 and HbF are reduced. Which of the following conditions is this patient likely to have
(A) Beta thalassemia minor
(B) Beta thalassemia major
(C) Alpha thalassemia trait
(D) Hydrops Fetalis
(E) Alpha thalassemia - HbH disease
E: The name implies it. The alpa haemaglobin gene contains two pairs on chromosome 16, accounting for 4 copies. Trait 1 and trait 2 results in one or two mutations. Trait 2 may be cis or trans. Mediterraneans and South East Asians tend to have the cis form, whilst trans is found more in Africa. HbH disease results from three mutations, with only one functional alpha gene. As a result, beta globins precipitate as beta 4. This leads to decreased production, increased haemolysis, target cells and HbH inclusions on peripheral film. Patients become transfusion dependent, demonstrate extramedullary haematopoiesis and may demonstrate iron overload. Hydrops fetalis results from mutations to all four alpha globulin genes and causes barts Hb = gamma 4. Beta thalassemia results in increased HbA2 concentrations and HbF concentrations.N Engl J Med 2014; 371:1908-1916
Question 15
Which condition results in the lowest level of hepcidin?
(A) Thalassemia intermedia
(B) Thalassemia major
(C) Thalassemia minor
(D) Anaemia of chronic disease
(E) Anaemia of renal failure
A: In thalassemia intermedia, patients are not transfused thus there is a high erythropoietic drive secondary to haemolysis and innefective erythropoiesis. The increased EPO results in decreased hepcidin. Thal major leads to secondary iron accumulation secondary to transfusional dependence. This increases iron levels, increases iron sensing and decreases hepatic synthesis of hepcidin. Hepcidin is increased in anaemia of chronic disease and anaemia of renal failure. This causes downregulation and internalisation of ferroportin therefore decreased Fe absorption and release of Fe from body stores. Origa et al, haematologica 2007
Question 16
Patients with beta-thalassaemia are at risk of infection. Which of the following infections is NOT associated with this condition
(A) Haemophilus
(B) Pneumococcal
(C) Yersinia
(D) Staphylococcal
(E) Meningococcal
D: Encapsulated organisms and yersinia (because it favours iron laden tissues) is particularly associated with beta-thalassaemia. RPA Course 2015
